![Image result for amg 3969]()
AMG-3969
M.Wt: 522.46
Cas : 1361224-53-4 , MF: C21H20F6N4O3S
WO 2012027261 PRODUCT PATENT
| Inventors |
Kate Ashton, Michael David Bartberger, Yunxin Bo, Marian C. Bryan, Michael Croghan, Christopher Harold Fotsch, Clarence Henderson Hale, Roxanne Kay Kunz, Longbin Liu, Nobuko Nishimura, Mark H. Norman, Lewis Dale Pennington, Steve Fong Poon, Markian Myroslaw Stec, Jean David Joseph St., Jr., Nuria A. Tamayo, Christopher Michael Tegley, Kevin Chao Yang |
| Applicant |
Amgen Inc. |
2-[4-[(2S)-4-[(6-Amino-3-pyridinyl)sulfonyl]-2-(1-propyn-1-yl)-1-piperazinyl]phenyl]-1,1,1,3,3,3-hexafluoro-2-propanol)
(S)-2-(4-(4-((6-Aminopyridin-3-yl)sulfonyl)-2-(prop-1-yn-1-yl)piperazin-1-yl)phenyl)-1,1,1,3,3,3-hexafluoropropan-2-ol,
mp 113–123 °C;
[α]D20 = +75.1 (c = 2.2, MeOH).
Agents for Type 2 Diabetes, PRECLINICAL
AMG-3969, a novel and stable small-molecule disruptor of glucokinase (GK) and glucokinase regulatory protein (GKRP) interaction by the optimization of initial screening hit and AMG-1694. AMG-3969 potently induced the dissociation of the GK-GKRP complex and promoted GK translocation both in-vitro and in-vivo. In rodent model of diabetes, AMG-3969 reduced blood glucose levels without affecting euglycemic animals. The study represents the first successful discovery of a small molecule that targets the GK-GKRP complex as a novel pathway for managing blood glucose levels with reduced hypoglycemic risk.
![Image result for AMGEN]()
![Kate Ashton]()
Kate Ashton
Senior Scientist at Amgen, Inc
Thousand Oaks, United States
Dr. Kate Ashton received a Masters in Chemistry with Industrial Experience from the University of Edinburgh. She conducted her PhD thesis research on the synthesis and structure elucidation of Reidispongiolide A with Prof. Ian Paterson at the University of Cambridge, and her postdoctoral work on SOMO catalysis with Prof. David W. C. MacMillan at both Caltech and Princeton. She has been at Amgen for 6 years and has worked on indications for cancer, Alzheimer’s and diabetes.Dr Fecke works in the area of industrial early drug discovery since 1996. He is currently Group Leader in the Primary Pharmacology department at UCB Pharma (UK) and is involved in the identification and characterization of NCE and NBE drugs in molecular interaction assays for both immunological and CNS diseases. Prior to joining UCB, he worked for Novartis and Siena Biotech in the areas of transplant rejection, neurodegeneration and oncology. He obtained his PhD at the Heinrich-Heine-University Dusseldorf in Germany in 1994.
![Image result for amg 3969]()
(S)-2-(4-(4-((6-Aminopyridin-3-yl)sulfonyl)-2-(prop-1-yn-1-yl)piperazin-1-yl)phenyl)-1,1,1,3,3,3-hexafluoropropan-2-ol, AMG-3969
Glucokinase (GK) is a member of a family of four hexokinases that are critical in the cellular metabolism of glucose. Specifically GK, also known as hexokinase IV or hexokinase D, facilitates glucose induced insulin secretion from pancreatic β-cells as well as glucose conversion into glycogen in the liver. GK has a unique catalytic activity that enables the enzyme to be active within the physiological range of glucose (from 5mM glucose to lOmM glucose).
Genetically modified mouse models support the role of GK playing an important role in glucose homeostasis. Mice lacking both copies of the GK gene die soon after birth from severe hyperglycemia, whereas mice lacking only one copy of the GK gene present with only mild diabetes. Mice that are made to overexpress the GK gene in their livers are hypoglycemic.
Numerous human mutations in the GK gene have been identified, with the vast majority of them resulting in proteins with impaired or absent enzymatic activity. These loss-of-function mutations are thought to contribute to the hyperglycemia seen with maturity-onset diabetes of the young type II (MODY-2). A small fraction of these mutations result in a GK with increased catalytic function. These individuals present with moderate to severe hypoglycemia.
GK activity in the liver is transiently regulated by glucokinase regulatory protein (GKRP). GK catalytic activity is inhibited when GK is bound to GKRP. This interaction is antagonized by increasing concentrations of both glucose and fructose -1 -phosphate (F1P). The complex of the two proteins is localized primarily to the nuclear compartment of a cell. Post prandially as both glucose and fructose levels rise, GK released from GKRP translocates to the cytoplasm. Cytoplasmic GK is now free of the inhibitory effects of GKRP and able to kinetically respond to glucose. Evidence from the Zucker diabetic fatty rat (ZDF) indicates that their glucose intolerance may be a result of this mechanism failing to function properly.
A compound that acts directly on GKRP to disrupt its interaction with GK and hence elevate levels of cytoplasmic GK is a viable approach to modulate GK activity. Such an approach would avoid the unwanted hypoglycemic effects of over stimulation of GK catalytic activity, which has been seen in the
development of GK activators. A compound having such an effect would be useful in the treatment of diabetes and other diseases and/or conditions in which GKRP and/or GK plays a role.
CLIP
Antidiabetic effects of glucokinase regulatory protein small-molecule disruptors
Nature 2013, 504(7480): 437
![Image result for Antidiabetic effects of glucokinase regulatory protein small-molecule disruptors.]()
![Image result for Antidiabetic effects of glucokinase regulatory protein small-molecule disruptors.]()
SYNTHESIS
![Figure]()
aReagents and conditions: (a) 1-propynylmagnesium bromide, THF, 0 °C, 99%; (b) TFA, DCM, then NaBH(OAc)3 77%; (c) NH4OH, EtOH, 120 °C, 88%; (d) chiral SFC, 38%………..Nature 2013,504, 437– 440
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012027261
EXAMPLE 241 : 2-(4-(4-((6-AMINO-3-PYRIDINYL)SULFONYL)-2-(l-PROP YN- 1 – YL)- 1 -PIPERAZINYL)PHENYL)- 1,1,1 ,3 ,3 ,3 -HEXAFLUORO-2-PROPANOL
![]()
STEP 1 : 4-BENZYL 1 -TERT-BUTYL 2-0X0-1,4-PIPERAZINEDICARBOXYLATE
A 2-L Erlenmeyer flask was charged with 2-piperazinone (36.5 g, 364 mmol, Sigma- Aldrich, St. Louis, MO), sodium carbonate (116 g, 1093 mmol), 600 mL of dioxane, and 150 mL of water. To this was slowly added benzyl chloroformate (62.1 g, 364 mmol, Sigma-Aldrich, St. Louis, MO) at room temperature over 20 min. After the addition was complete, the mixture was stirred for 2 h and then diluted with water and extracted with EtOAc (2 L). The combined organic extracts were dried (MgS04), filtered, and concentrated to give a white solid. To this solid was added 500 mL of DCM, triethylamine (128 mL, 911 mmol), DMAP (4.45 g, 36.4 mmol), and di-tert-butyl dicarbonate (119 g, 546 mmol, Sigma-Aldrich, St. Louis, MO). After 1 h at room temperature, the mixture was diluted with water and the organics were separated. The organics were dried (MgS04), filtered, and concentrated to give a brown oil. To this oil was added 100 mL of DCM followed by 1 L of hexane. The resulting white solid was collected by filtration to give 4-benzyl 1-tert-butyl 2-oxo-l,4-piperazinedicarboxylate (101 g).
STEP 2: BENZYL (2-((TERT-BUTOXYCARBONYL)AMINO)ETHYL)(2-OXO-3 -PENTYN- 1 -YL)CARBAMATE
A 150-mL round-bottomed flask was charged with 4-benzyl 1-tert-butyl
2- oxo-l,4-piperazinedicarboxylate (1.41 g, 4.22 mmol) and THF (5 mL). 1-Propynylmagnesium bromide (0.5 M in THF, 20.0 mL, 10.0 mmol, Sigma-Aldrich, St. Louis, MO) was added at 0 °C slowly. The mixture was stirred at 0 °C for 2 h. Saturated aqueous NH4C1 (40 mL) was added and the aqueous phase was extracted with EtOAc (200 mL, then 2 x 100 mL). The combined organic phases were dried over sodium sulfate, filtered and concentrated in vacuo. The crude product was purified by column chromatography (50 g of silica, 0 to 50% EtOAc in hexanes) to afford benzyl (2-((tert-butoxycarbonyl)amino)ethyl)(2-oxo- 3- pentyn-l-yl)carbamate (1.55 g) as a clear oil.
STEP 3: BENZYL 3-(l-PROPYN-l-YL)-l-PIPERAZINECARBOXYLATE
A 3-L round-bottomed flask was charged with 2-((tert-butoxycarbonyl)amino)ethyl)(2-oxo-3-pentyn-l-yl)carbamate (82.2 g, 219 mmol) and 300 mL of DCM. After cooling to -10 °C, TFA (169 mL, 2195 mmol) was added and the resulting dark solution was stirred at room temperature for 15 min. Sodium triacetoxyborohydride (186 g, 878 mmol, Sigma-Aldrich, St. Louis, MO) was then added portion- wise over 10 min. After 2 h, the mixture was
concentrated, diluted with EtOAc (1 L), and neutralized with 5 N NaOH. The layers were separated and the organic extracts were washed with brine, dried (MgS04), filtered and concentrated. The resulting orange oil was purified via column chromatography (750 g of silica gel, 0 to 4.5 % MeOH/DCM) to give benzyl 3-(l-propyn-l-yl)-l-piperazinecarboxylate (43.7 g) as a brown foam.
STEP 4: BENZYL 3-(l-PROPYN-l-YL)-4-(4-(2,2,2-TRIFLUORO-l-HYDROXY- 1 -(TRIFLUOROMETHYL)ETHYL)PHENYL)- 1 -PIPERAZINECARBOXYLATE
A 150-mL reaction vessel was charged with benzyl 3-(prop-l-yn-l-yl)piperazine-l-carboxylate (2.88 g, 11.2 mmol), 2-(4-bromophenyl)-l, 1,1, 3,3,3-hexafluoropropan-2-ol (4.36 g, 13.5 mmol, Bioorg. Med. Chem. Lett. 2002, 12, 3009), dicyclohexyl(2′,6′-diisopropoxy-[ 1 , 1 ‘-biphenyl]-2-yl)phosphine, RuPhos (0.530 g, 1.14 mmol, Sigma- Aldrich, St. Louis, MO), RuPhos Palladacycle (0.417 g, 0.572 mmol, Strem Chemical Inc, Newburyport, MA), sodium tert-butoxide (2.73 g, 28.4 mmol, Strem Chemical Inc, Newburyport, MA) and toluene (35 mL). The mixture was degassed by bubbling Ar through the solution for 10 min. The vessel was sealed and heated at 100 °C for 1.5 h. The reaction mixture was cooled to room temerature and water (100 mL) was added. The aqueous phase was extracted with EtOAc (3 x 100 mL) and the combined organic phases were washed with saturated aqueous sodium chloride (150 mL). The organic extracts were dried over sodium sulfate, filtered and concentrated in vacuo. The crude product was purified by column chromatography (100 g of silica, 0 to 50% EtOAc in hexanes) to afford benzyl 3-(l-propyn-l-yl)-4-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -(trifluoromethyl)ethyl)phenyl)- 1 -piperazinecarboxylate as a yellow solid.
STEP 5: 2-(4-(4-((6-CHLORO-3-PYRIDINYL)SULFONYL)-2-(l-PROPYN-l-YL)- 1 -PIPERAZIN YL)PHENYL)- 1,1,1 ,3 ,3 ,3 -HEXAFLUORO-2-PROPANOL
A 500-mL round-bottomed flask was charged with benzyl 3-(l-propyn-l-yl)-4-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -(trifluoromethyl)ethyl)phenyl)- 1 -piperazinecarboxylate (3.13 g, 6.25 mmol) and TFA (40 mL).
Trifluoromethanesulfonic acid (1.25 mL, 14.1 mmol, Acros/Fisher Scientific, Waltham, MA) was added dropwise at room temperature. After 5 min, additional TfOH (0.45 mL, 5.1 mmol) was added. After an additional 10 min, solid
NaHC03 was carefully added in potions. Saturated aqueous NaHC03 (250 mL) was added slowly to bring pH to approximately 7. The aqueous phase was extracted with EtOAc (100 mL). At this time, more solid NaHC03 was added to the aqueous phase and extracted again with EtOAc (100 mL). The combined organic phases were washed with water (200 mL) and saturated aqueous sodium chloride (200 mL). The combined organic extracts were dried over sodium sulfate, filtered and concentrated in vacuo to afford 3.10 g of tan solid.
A 500-mL round-bottomed flask was charged with this material, triethylamine (5.00 mL, 35.9 mmol) and CH2CI2 (30 mL). 6-Chloropyridine-3-sulfonyl chloride (1.58 g, 7.43 mmol, Organic Process Research & Development 2009, 13, 875) was added in potions at 0 °C. The brown mixture was stirred at 0 °C for 10 min. The volume of the reaction mixture was reduced to approximately 10 mL in vacuo then the mixture was purified twice by column chromatography (100 g of silica, 0 to 50% EtOAc in hexanes) to afford 2-(4-(4-((6-chloro-3-pyridinyl)sulfonyl)-2-( 1 -propyn- 1 -yl)- 1 -piperazinyl)phenyl)- 1,1,1,3,3,3-hexafluoro-2-propanol (3.46 g) as an off-white solid.
STEP 6: 2-(4-(4-((6-AMINO-3-PYRIDINYL)SULFONYL)-2-(l-PROPYN-l-YL)- 1 -PIPERAZIN YL)PHENYL)- 1,1,1 ,3 ,3 ,3 -HEXAFLUORO-2-PROPANOL
A 20-mL sealed tube was charged with 2-(4-(4-((6-chloro-3-pyridinyl)sulfonyl)-2-( 1 -propyn- 1 -yl)- 1 -piperazinyl)phenyl)- 1,1,1,3,3,3-hexafluoro-2-propanol (0.340 g, 0.627 mmol), concentrated ammonium hydroxide (5.00 mL, 38.5 mmol) and EtOH (5 mL). The reaction mixture was heated in an Initiator (Biotage, AB, Uppsala, Sweden) at 120 °C for 1 h. The reaction mixture was further heated in a heating block at 110 °C for 5 h. The reaction mixture was concentrated and purified by column chromatography (25 g of silica, 30 to 80% EtOAc in hexanes) to afford 2-(4-(4-((6-amino-3-pyridinyl)sulfonyl)-2-( 1 -propyn- 1 -yl)- 1 -piperazinyl)phenyl)- 1,1,1,3,3,3-hexafluoro-2-propanol (0.289 g) as a mixture of two enantiomers.
1H NMR (400 MHz, CDC13) δ ppm 8.49 (br. s., 1 H), 7.80 (dd, J= 2.3, 8.8 Hz, 1 H), 7.59 (d, J= 8.8 Hz, 2 H), 6.97 (d, J= 9.0 Hz, 2 H), 6.55 (d, J= 8.8 Hz, 1 H), 5.05 (s, 2 H), 4.46 (br. s., 1 H), 3.85 – 3.72 (m, 2 H), 3.54 (br. s., 1 H), 3.50 – 3.34 (m, 2 H), 2.83 (dd, J= 3.3, 11.0 Hz, 1 H), 2.69 (dt, J= 3.4, 11.0 Hz, 1 H), 1.80 (s, 3 H). m/z (ESI, +ve ion) 523.1 (M+H)+. GK-GKRP IC50 (Binding) = 0.003 μΜ
The individual enantiomers were isolated using chiral SFC. The method used was as follows: Chiralpak® ADH column (21 x 250 mm, 5 μιη) using 35% methanol in supercritical C02 (total flow was 70 mL/min). This produced the two enantiomers with enantiomeric excesses greater than 98%.
![]()
2-(4-((2S)-4-((6-amino-3-pyridinyl)sulfonyl)-2-(l -propyn- 1-yl)- 1 -piperazinyl)phenyl)- 1,1,1 ,3 ,3 ,3 -hexafluoro-2-propanol and 2-(4-((2R)-4-((6-amino-3 -pyridinyl)sulfonyl)-2-( 1 -propyn- 1 -yl)- 1 -piperazinyl)phenyl)- 1,1,1,3,3,3-hexafluoro-2-propanol.
FIRST ELUTING PEAK (PEAK #1)
1H NMR (400 MHz, CDC13) δ 8.48 (d, J= 2.3 Hz, 1 H), 7.77 (dd, J= 2.5, 8.8 Hz, 1 H), 7.57 (d, J= 8.8 Hz, 2 H), 6.95 (d, J= 9.2 Hz, 2 H), 6.52 (d, J= 8.8 Hz, 1 H), 4.94 (s, 2 H), 4.44 (br. s., 1 H), 3.82 – 3.71 (m, 2 H), 3.58 – 3.33 (m, 3 H), 2.81 (dd, J= 3.2, 11.1 Hz, 1 H), 2.67 (dt, J= 3.9, 11.0 Hz, 1 H), 1.78 (d, J = 2.2 Hz, 3 H). m/z (ESI, +ve ion) 523.2 (M+H)+. GK-GKRP IC50 (Binding) = 0.002 μΜ.
SECOND ELUTING PEAK (PEAK #2)
1H NMR (400 MHz, CDC13) δ 8.49 (d, J= 1.8 Hz, 1 H), 7.78 (dd, J= 2.3, 8.8 Hz, 1 H), 7.59 (d, J= 8.6 Hz, 2 H), 6.97 (d, J= 9.0 Hz, 2 H), 6.54 (d, J= 8.8 Hz, 1 H), 4.97 (s, 2 H), 4.46 (br. s., 1 H), 3.77 (t, J= 11.7 Hz, 2 H), 3.67 (br. s., 1 H), 3.51 – 3.33 (m, 2 H), 2.82 (dd, J= 3.3, 11.0 Hz, 1 H), 2.68 (dt, J= 3.9, 11.1 Hz, 1 H), 1.79 (d, J= 2.0 Hz, 3 H). m/z (ESI, +ve ion) 523.2 (M+H)+. GK-GKRP IC50 (Binding) = 0.342 μΜ.
Alternative procedure starting after Step 4.
![]()
STEP 5 ‘: 2-(4-(4-((6-AMINO-3-PYRIDINYL)SULFONYL)-2-(l-PROPYN-l-YL)- 1 -PIPERAZIN YL)PHENYL)- 1,1,1 ,3 ,3 ,3 -HEXAFLUORO-2-PROPANOL
Alternatively, 2-(4-(4-((6-amino-3-pyridinyl)sulfonyl)-2-( 1 -propyn- 1 -yl)-l-piperazinyl)phenyl)-l,l,l,3,3,3-hexafluoro-2-propanol was synthesized from benzyl 3-( 1 -propyn- 1 -yl)-4-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -(trifluoromethyl)ethyl)phenyl)- 1 -piperazinecarboxylate as follows.
A 2-L round-bottomed flask was charged with benzyl 3 -(1 -propyn- 1-yl)-4-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -(trifluoromethyl)ethyl)phenyl)- 1 -piperazinecarboxylate (21.8 g, 43.5 mmol, step 5) and TFA (130 mL).
Trifluoromethanesulfonic acid (11.6 mL, 131 mmol, Acros/Fisher Scientific, Waltham, MA) was added slowly at rt resulting orange cloudy mixture. After stirring at rt for 10 min, the volume of the reaction mixture was reduced to half in vacuo. Solid NaHC03 was added in potions until the mixture became sludge. Saturated aqueous NaHC03(800 mL) was added slowly until the pH was about
8. The aqueous phase was extracted with EtOAc (3 x 250 mL). The combined organic phases were washed with water (500 mL) and saturated aqueous NaCl (500 mL). The organic phase was dried over sodium sulfate, filtered and concentrated in vacuo. This material was dissolved into DCM (200 mL) and triethylamine (31.0 mL, 222 mmol) was added. Then 6-aminopyridine-3-sulfonyl chloride (9.40 g, 48.8 mmol, published PCT patent application no. WO
2009/140309) was added in potions over 10 min period. The brown mixture was stirred at room temperature for 10 min. The reaction mixture was washed with water (300 mL) and saturated aqueous NaCl (300 mL). The organic phase was dried over sodium sulfate, filtered and concentrated in vacuo. The crude product was purified by column chromatography (780 g of total silica, 30 to 90% EtOAc in hexanes) to afford 2-(4-(4-((6-amino-3-pyridinyl)sulfonyl)-2-(l-propyn-l-yl)-l-piperazinyl)phenyl)-l,l,l,3,3,3-hexafluoro-2-propanol (19.4 g) as a mixture of two enantiomers.
Paper
Nonracemic Synthesis of GK–GKRP Disruptor AMG-3969
† Therapeutic Discovery, Amgen Inc., One Amgen Center Drive, Thousand Oaks, California 91320, United States
‡ Amgen Inc. 360 Binney Street, Cambridge, Massachusetts 02142, United States
J. Org. Chem., 2014, 79 (8), pp 3684–3687
![Abstract Image]()
A nonracemic synthesis of the glucokinase–glucokinase regulatory protein disruptor AMG-3969 (5) is reported. Key features of the synthetic approach are an asymmetric synthesis of the 2-alkynyl piperazine core via a base-promoted isomerization and a revised approach to the synthesis of the aminopyridinesulfonamide with an improved safety profile.
(S)-2-(4-(4-((6-Aminopyridin-3-yl)sulfonyl)-2-(prop-1-yn-1-yl)piperazin-1-yl)phenyl)-1,1,1,3,3,3-hexafluoropropan-2-ol, AMG-3969 (5)
(
S)-2-(4-(4-((6-aminopyridin-3-yl)sulfonyl)-2-(prop-1-yn-1-yl)piperazin-1-yl)phenyl)-1,1,1,3,3,3-hexafluoropropan-2-ol (
5) (64.0 g, 49% yield) as white solid. The enanatiomeric excess was found to be >99.5% by chiral SFC (see
Supporting Information):
1H NMR (400 MHz, CDCl3) δ 8.47 (s, 1 H), 7.79 (d, J = 8.6 Hz, 1 H), 7.59 (d, J = 8.2 Hz, 2 H), 6.97 (d, J = 8.6 Hz, 2 H), 6.55 (d, J = 8.8 Hz, 1 H), 5.06 (br s, 2 H), 4.45 (br s, 1 H), 3.96 (br s, 1 H), 3.77 (t, J = 12.1 Hz, 2 H), 3.50–3.35 (m, 2 H), 2.82 (d, J = 11.0 Hz, 1 H), 2.68 (t, J = 10.9 Hz, 1 H), 1.79 (s, 3 H);
13C NMR (101 MHz, CD3OD) δ 163.8, 152.0, 150.1, 138.2, 129.0, 124.7 (q), 123.9, 121.1, 117.5, 109.3, 82.8, 78.3 (m), 75.5, 52.0, 47.2, 44.9, 3.2;
HRMS (ESI-TOF) m/z [M + H]+calcd for C21H21F6N4O3S 523.1239, found 523.1229;
mp 113–123 °C;
[α]D20 = +75.1 (c = 2.2, MeOH).
Clip
![]()
AMG-3969 is a disruptor of the glucokinase (GK)–glucokinase regulatory protein (GKRP) protein–protein interaction. Bourbeau and co-workers at Amgen describe their efforts towards an asymmetric synthesis of this compound ( J. Org. Chem. 2014, 79, 3684). The discovery route to this compound involved seven steps (14% overall yield), had certain safety concerns and relied upon SFC separation of the API enantiomers. The new route requires five steps (26% overall yield) and delivers the API in excellent enantiomeric excess (99% ee). A key feature of the synthetic approach was an asymmetric synthesis of the 2-alkynylpiperazine core via a base-promoted isomerization. It was found that the strongly basic conditions employed for the “alkyne-walk” did not erode the previously established stereocenter. Also, safety concerns around a late-stage amination of a 2-chloropyridine intermediate in the discovery route were alleviated by starting with a Boc-protected diaminopyridine instead.
PATENT
INTERMEDIATE A: TERT-EUTYL (5-(CHLOROSULFONYL)-2-PYRIDINYL)CARBAMATE
0,N
![]()
STEP 1 : TERT-BUTY (5-NITRO-2-PYRIDINYL)CARBAMATE
A 3-L round-bottomed flask was charged with 5-nitro-2-pyridinamine (75.0 g, 539 mmol, Alfa Aesar, Ward Hill, MA) and 500 mL of DCM. To this was added triethylamine (82 g, 810 mmol), di-tert-butyl dicarbonate (129 g, 593 mmol, Sigma-Aldrich, St. Louis, MO), and N,N-dimethylpyridin-4-amine (32.9 g, 270 mmol, Sigma-Aldrich, St. Louis, MO). After stirring at rt for 18 h, the mixture was diluted with water and the solid was collected by filtration. The yellow solid was washed with MeOH to give tert-butyl (5-nitro-2-pyridinyl)carbamate (94.6 g) as a light yellow solid.
STEP 2: TERT-BUTY (5 – AMINO-2-P YRIDINYL)C ARB AM ATE
A 3-L round-bottomed flask was charged with tert-butyl (5-nitro-2-pyridinyl)carbamate (96.4 g, 403 mmol), 500 mL of MeOH, 500 mL of THF, and 100 mL of sat aq NH4Cl. Zinc (105 g, 1610 mmol, Strem Chemical Inc, Newburyport, MA) was slowly added (over 10 min) to this solution. The mixture was stirred at room temperature for 12 h, then filtered. The filtrate was concentrated and then diluted with EtOAc and washed with water. The organic extracts were dried over MgS04, filtered, and concentrated. The resulting solid was recrystallized from MeOH to give tert-butyl(5-amino-2-pyridinyl)carbamate (38.6 g) as a light-yellow solid.
STEP 3: TERT-BUTYL (5-(CHLOROSULFONYL)-2-PYRIDINYL)CARBAMATE
A 3-L round-bottomed flask was charged with sodium nitrite (15.3 g, 221 mmol, J. T. Baker, Philipsburg, NJ), 100 mL of water and 500 mL of MeCN. After cooling to 0 °C, cone, hydrochloric acid (231 mL, 2770 mmol) was slowly added keeping the internal temperature below 10 °C. After stirring at 0 °C for 10 min, tert-butyl (5-amino-2-pyridinyl)carbamate (38.6 g, 184 mmol) was added as a suspension in MeCN (200 mL). The mixture was stirred for 30 min, then 150 mL of AcOH, copper(ii) chloride (12.4 g, 92.2 mmol, Sigma-Aldrich, St. Louis, MO), and copper(i) chloride (0.183 g, 1.85 mmol, Strem Chemical Inc,
Newburyport, MA) were added. S02 gas (Sigma-Aldrich, St. Louis, MO) was bubbled through the solution for 15 min. The mixture was stirred at 0 °C for 30 min, then about 500 mL of ice-cold water was added. The resulting precipitate was collected by filtration and dried over MgS04 to give tert-butyl (5-(chlorosulfonyl)-2-pyridinyl)carbamate (15.5 g) as a white solid.
1H NMR (400MHz, CDC13) δ ppm 8.93 (br s, 1 H), 8.63 – 8.42 (m, 1 H), 8.35 -7.94 (m, 2 H), 1.58 (s, 9 H).
INTERMEDIATE B: (3S)-l-BENZYL-3-(l-PROPYN-l-YL)PIPERAZINE
![]()
STEP 1 : (3S)-l-BENZYL-3-(2-PROPYN-l-YL)-2,5-PIPERAZINEDIONE
A 1-L round-bottoemd flask was charged with (S)-2-((tert-butoxycarbonyl)amino)pent-4-ynoic acid (42.0 g, 197 mmol, AK Scientific, Union City, CA), ethyl 2-(benzylamino)acetate (40.0 g, 207 mmol, Sigma-Aldrich, St. Louis, MO), HATU (90 g, 240 mmol, Oakwood Products, West Columbia, SC) and 200 mL of DMF. To this was added N-ethyl-N-isopropylpropan-2-amine (51.5 ml, 296 mmol, Sigma-Aldrich, St. Louis, MO). After 15 min of stirring at rt, the mixture was diluted with water 300 mL and extracted with 1 L of 20% EtOAc in diethyl ether. The layers were separated and the organic was washed with 2 M HCl, water, sat. aq. NaHC03 and brine. The extracts were dried and concentrated to give an off-white solid. To this was added 200 mL of DCM and TFA (152 ml, 1970 mmol, Sigma-Aldrich, St. Louis, MO). After stirring at rt for 30 min, the mixture was concentrated and then azetroped with 100 mL toluene (twice). To the brown oil obtained was added ammonia (2 M in MeOH, 394 ml, 789 mmol, Sigma-Aldrich, St. Louis, MO). The mixture was stirred at rt for 30 min. The mixture was concentrated, dissolved in EtOAc, and washed with water. The organics were dried (MgS04), filtered, and concentrated to give a white solid that was triturated with diethyl ether to give (S)-l-benzyl-3-(prop-2-yn-l-yl)piperazine-2,5-dione (37.3 g) as a white solid.
STEP 2: (3S)-l-BENZYL-3-(2-PROPYN-l-YL)PIPERAZINE
A 1-L round-bottomed flask was charged with (S)-l-benzyl-3-(prop-2-yn-l-yl)piperazine-2,5-dione (37.3 g, 154 mmol) and 150 mL of THF. To this was slowly added aluminum (III) lithium hydride (1M in THF, 539 ml, 539 mmol, Sigma-Aldrich, St. Louis, MO). After the addition was complete the mixture was heated at 80 °C for 12 h. The mixture was then cooled to 0 °C and solid sodium sulfate decahydrate was added until bubbling ceased. The mixture was filtered and the filtrate was concentrated to give (S)-l-benzyl-3-(prop-2-yn-l-yl)piperazine (18.1 g) as a yellow oil.
STEP 3: (35)-l-BENZYL-3-(l-PROPYN-l-YL)PIPERAZINE
To a solution of (35)-l-benzyl-3-(2-propyn-l-yl)piperazine (2.3 g, 11 mmol) in THF (50 mL) was added potassium t-butoxide (2.41 g, 21.5 mmol, Sigma-Aldrich, St. Louis, MO). The reaction mixture was stirred at rt for 30 min, then quenched with water (200 mL) and EtOAc (300 mL) was added. The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum to give a solid that was purified by silica gel column chromatography (0 to 10% MeOH in CH2CI2) and then recrystallized from hexanes to afford (35)- 1-benzyl-3-(l-propyn-l-yl)piperazine (2.16 g) as an off-white solid.
1H NMR (400MHz, CD3OD) δ ppm 7.42 – 7.21 (m, 5 H), 3.59 – 3.49 (m, 3 H), 2.93 (td, J= 2.9, 12.4 Hz, 1 H), 2.86 – 2.73 (m, 2 H), 2.68 (d, J= 11.3 Hz, 1 H), 2.22 – 2.04 (m, 2 H), 1.80 (d, J= 2.3 Hz, 3 H).
INTERMEDIATE C: N,N-BIS(4-METHOXYBENZYL)-5-(((35)-3-(l-PROPYN- 1 – YL)- 1 -PIPERAZINYL)SULFONYL)-2-PYRIDIN AMINE
![]()
STEP 1 : (35)-l-((6-CHLORO-3-PYRIDINYL)SULFONYL)-3-(l-PROPYN-l-YL)PIPERAZINE
To a stirred solution of benzyl (35)-3-(l-propyn-l-yl)-l-piperazinecarboxylate (2.51 g, 9.71 mmol, Intermediate E) in TFA (20 mL) in 250-mL round-bottomed flask, trifluoromethanesulfonic acid (2.59 mL, 29.1 mmol, Alfa Aesar, Ward Hill, MA) was added slowly at rt. After stirring at room temperature for 3 min, the reaction mixture was concentrated to dryness under a vacuum. DCM (20 mL) was added to the residue followed by triethylamine (13.5 mL, 97 mmol). After the material went into solution, the mixture was cooled to 0 °C and 6-chloro-3-pyridinesulfonyl chloride (2.06 g, 9.73 mmol, Organic Process Research & Development 2009, 13, 875) was added portion-wise. After 5 min of stirring at 0 °C, water (40 mL) was added at that temperature and the layers were separated. The aqueous phase was extracted with DCM (2 x 50 mL). The combined organic phases were washed with saturated aqueous sodium chloride (60 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under a vacuum. The crude product was purified by column chromatography (100 g of silica, 30 to 90% EtOAc in hexanes) to afford (35)- 1-((6-chloro-3-pyridinyl)sulfonyl)-3-(l-propyn-l-yl)piperazine (2.61 g) as an off-white solid.
STEP 2: N,N-BIS(4-METHOXYBENZYL)-5-(((35)-3-(l-PROPYN-l-YL)-l-PIPERAZINYL)SULFONYL)-2-PYRIDIN AMINE
A mixture of (35)-l-((6-chloro-3-pyridinyl)sulfonyl)-3-(l-propyn-l-yl)piperazine (2.6 g, 8.7 mmol), N-(4-methoxybenzyl)-l-(4-methoxyphenyl)methanamine (2.40 g, 9.33 mmol, WO2007/109810A2), and DIPEA (2.4 mL, 14 mmol) in z‘-BuOH (8.0 mL) was heated at 132 °C using a microwave reactor for 3 h. This reaction was run three times (total starting material amount was 7.2 g). The mixtures from the three runs were combined and partitioned between EtOAc (200 mL) and aqueous NaHC03 (half saturated, 50 mL). The organic layer was washed with aqueous NaHC03 (3 x 50 mL), dried over Na2S04, filtered, and concentrated. The residue was purified (5-times total) by chromatography on silica using MeOH:DCM:EtOAc:hexane
(4:20:20:60) as eluent to give N,N-bis(4-methoxybenzyl)-5-(((3S)-3-(l-propyn-i-yl)-l-piperazinyl)sulfonyl)-2-pyridinamine (6.6 g) as a white foam.
1H NMR (400MHz ,CDC13) δ ppm 8.55 (d, J= 2.3 Hz, 1 H), 7.64 (dd, J= 2.5, 9.0 Hz, 1 H), 7.13 (d, J= 8.6 Hz, 4 H), 6.91 – 6.81 (m, 4 H), 6.47 (d, J= 9.0 Hz, 1 H), 4.75 (s, 4 H), 3.80 (s, 6 H), 3.68 – 3.61 (m, 1 H), 3.57 (d, J= 11.2 Hz, 1 H), 3.41 (d, J= 11.3 Hz, 1 H), 3.07 (td, J= 3.3, 12.1 Hz, 1 H), 2.87 (ddd, J= 2.9, 9.7, 12.2 Hz, 1 H), 2.63 – 2.47 (m, 2 H), 1.80 (d, J= 2.2 Hz, 3 H). One exchangeable proton was not observed, m/z (ESI, +ve ion) 521.2 (M+H)+.
INTERMEDIATE D: rEi?r-BUTYL(5-(((35)-3-(l-PROPYN-l-YL)-4-(4-(2-(TRIFLUOROMETHYL)-2-OXIRANYL)PHENYL)- 1 -PIPERAZINYL)SULFONYL)-2-PYRIDINYL)CARBAMATE
![]()
step 1 step 2
![]()
STEP 1 : l-BR0M0-4-(l-(TRIFLU0R0METHYL)ETHENYL)BENZENE
To a 1-L round-bottomed flask was added methyl phenylphosphonium bromide (25.4 g, 71.1 mmol, Sigma- Aldrich, St. Louis, MO) and toluene (75 mL). The resulting mixture was stirred for 5 min then concentrated and dried under high vacuum for 30 min. To this residue was added THF (300 mL) followed by n-butyllithium (2.5 M in hexanes, 29.0 mL, 71.1 mmol, Aldrich, St. Louis, MO) dropwise via an addition funnel. After being stirred for 1 h at rt, a solution of l-(4-bromophenyl)-2,2,2-trifluoroethanone (15.0 g, 59.3 mmol, Matrix Scientific, Columbia, SC) in THF (20 mL) was added to the reaction mixture dropwise via an addition funnel. The reaction mixture was stirred at rt for 2 h. The reaction was quenched with saturated aqueous NH4C1 and the mixture was concentrated. The residue was partitioned between diethyl ether (150 mL) and saturated aqueous NH4C1 (80 mL). The organic layer was washed with water and brine, dried over MgS04, filtered, and concentrated. The resulting crude product was purified by column chromatography (330 g of silica gel, 2 to 5% EtOAc in hexanes) to afford l-bromo-4-(l-(trifluoromethyl)ethenyl)benzene (14.0 g) as a brown liquid.
STEP 2: 2-(4-BROMOPHENYL)-3,3,3-TRIFLUORO-l,2-PROPANEDIOL
To a solution of l-bromo-4-(l-(trifluoromethyl)ethenyl)benzene (13.5 g, 53.8 mmol) in acetone (100 mL) and water (100 mL) was added NMO (6.90 g, 59.2 mmol, Sigma- Aldrich, St. Louis, MO) and osmium tetroxide (0.140 mL, 2.70 mmol, Sigma-Aldrich, St. Louis, MO). The resulting mixture was stirred at rt for 6 h. The reaction mixture was filtered and the filtrate was concentrated. The residue was partitioned between EtOAc (100 mL) and water (30 mL). The aqueous layer was extracted with EtOAc (2 x 75 mL). The combined organic layers were dried over MgS04, filtered, and concentrated. The resulting product was purified by column chromatography (330 g of silica gel, 0 to 8% MeOH in DCM) to afford 2-(4-bromophenyl)-3,3,3-trifluoro-l,2-propanediol (14.5 g) as an off-white solid.
STEP 3: 4-(4-BROMOPHENYL)-2,2-DIMETHYL-4-(TRIFLUOROMETHYL)-1,3-DIOXOLANE
To a solution of 2-(4-bromophenyl)-3,3,3-trifluoro-l,2-propanediol (14.5 g, 51.0 mmol) in acetone (200 mL) was added 2,2-dimethoxypropane (19.0 mL, 153 mmol, Sigma-Aldrich, St. Louis, MO) and /?-toluenesulfonic acid (0.485 g, 2.54 mmol, Sigma-Aldrich, St. Louis, MO). The resulting mixture was stirred at rt for 20 h. Additional 2,2-dimethoxypropane (19.0 mL, 153 mmol, Sigma-Aldrich, St. Louis, MO) and /?-toluenesulfonic acid (0.485 g, 2.54 mmol, Sigma-Aldrich, St. Louis, MO) were added and the reaction was stirred for another 20 h. The reaction was quenched with saturated aqueous NaHC03 (10 mL). The reaction mixture was concentrated and the residue was partitioned between
EtOAc (100 mL) and saturated aqueous NaHC03 (60 mL). The aqueous layer was extracted with EtOAc (2 x 50 mL). The combined organic layers were dried over MgS04, filtered, and concentrated. The resulting product was purified by column chromatography (330 g of silica gel, 0 to 8% EtOAc in hexanes) to afford 4-(4-bromophenyl)-2,2-dimethyl-4-(trifluoromethyl)-l,3-dioxolane (15.7 g) as a colorless liquid.
STEP 4: BENZYL (3S)-4-(4-(2,2-DIMETHYL-4-(TRIFLUOROMETHYL)-l,3-DIOXOLAN-4-YL)PHENYL)-3-(l -PROPYN- 1 -YL)- 1 -PIPERAZINECAPvBOXYLATE
To a 20-mL vial was added benzyl (3S)-3-(l -propyn- l-yl)-l-piperazinecarboxylate (1.0 g, 3.87 mmol, Intermediate E), RuPhos Palladacycle (0.250 g, 0.310 mmol, Strem Chemical, Newburyport, MA), 4-(4-bromophenyl)-2,2-dimethyl-4-(trifluoromethyl)-l,3-dioxolane (2.50 g, 7.74 mmol), dioxane (15.0 mL), and sodium t-butoxide (0.740 g, 7.74 mmol, Sigma-Aldrich, St.
Louis, MO). The reaction mixture was degassed by bubbling N2 through the solution for 5 min, then the vial was capped. The reaction mixture was heated at 80 °C for 30 min then allowed to cool to rt and partitioned between EtOAc (70 mL) and water (40 mL). The aqueous layer was extracted with EtOAc (1 x 50 mL). The combined organic layers were dried over MgS04, filtered, and concentrated. The crude product was purified by column chromatography (80 g of silica, 5% to 30% EtOAc in hexanes) to afford benzyl (35)-4-(4-(2,2-dimethyl-4-(trifluoromethyl)- 1 ,3-dioxolan-4-yl)phenyl)-3-(l -propyn- 1 -yl)- 1 -piperazinecarboxylate (1.6 g) as a yellow foam.
STEP 5: rEi?r-BUTYL(5-(((35)-3-(l-PROPYN-l-YL)-4-(4-(2,2,2-TRIFLUORO- 1 -HYDROXY- 1 -(HYDROXYMETH YL)ETHYL)PHENYL)- 1 -PIPERAZINYL)SULFONYL)-2-PYRIDINYL)CARBAMATE
To a 150-mL round-bottomed flask was added benzyl (3S)-4-(4-(2,2-dimethyl-4-(trifluoromethyl)- 1 ,3 -dioxolan-4-yl)phenyl)-3 -( 1 -propyn- 1 -yl)- 1 -piperazinecarboxylate (1.60 g, 3.18 mmol) and TFA (20 mL, Sigma-Aldrich, St. Louis, MO). After the substrate was completely dissolved in TFA,
trifluoromethanesulfonic acid (0.850 mL, 9.55 mmol, Alfa Aesar, Ward Hill,
MA) was added and the resulting mixture was stirred at rt for 1.5 h. The reaction mixture was slowly poured into a 300-mL beaker which contained 100 mL ice water. The resulting mixture was stirred while NaOH pellets (11.0 g) were slowly added to adjust the pH to 7. The solution was extracted with EtOAc (2 x 70 mL) and 10% IPA in CHCI3 (2 x 40 mL). The combined organic layers were dried over MgS04, filtered, and concentrated. The resulting intermediate was redissolved in DCM (60 mL). Triethylamine (2.20 mL, 16.0 mmol, Sigma-Aldrich, St. Louis, MO) and tert-butyl (5-(chlorosulfonyl)-2-pyridinyl)carbamate (1.04 g, 3.60 mmol, Intermediate A) were added. The reaction mixture was stirred at rt for 1 h then partitioned between DCM (70 mL) and water (30 mL). The aqueous layer was extracted with DCM (2 x 40 mL). The combined organic layers were dried over MgS04, filtered, and concentrated. The crude product was purified by column chromatography (120 g of silica, 10% to 40% acetone in hexanes) to afford tert-butyl (5-(((35)-3-(l-propyn-l-yl)-4-(4-(2,2,2-trifiuoro-l-hydroxy- 1 -(hydroxymethyl)ethyl)phenyl)- 1 -piperazinyl)sulfonyl)-2-pyridinyl)carbamate (1.0 g) as a yellow foam.
STEP 6: rEi?r-BUTYL(5-(((35)-3-(l-PROPYN-l-YL)-4-(4-(2-(TRIFLUOROMETHYL)-2-OXIRANYL)PHENYL)- 1 -PIPERAZINYL)SULFONYL)-2-PYRIDINYL)CARBAMATE
To a solution of tert-butyl (5-(((35)-3-(l-propyn-l-yl)-4-(4-(2,2,2-trifiuoro- 1 -hydroxy- 1 -(hydroxymethyl)ethyl)phenyl)- 1 -piperazinyl)sulfonyl)-2-pyridinyl)carbamate (0.300 g, 0.513 mmol) in DCM (5 mL) was added triethylamine (0.400 mL, 2.88 mmol, Sigma-Aldrich, St. Louis, MO) and p-toluenesulfonyl chloride (0.108 g, 0.564 mmol, Sigma-Aldrich, St. Louis, MO). The resulting mixture was heated at reflux (50 °C) under N2 for 2 h. The reaction mixture was cooled to rt and partitioned between sat. NaHCOs (30 mL) and DCM (70 mL). The aqueous layer was extracted with DCM (2 x 40 mL). The combined organic layers were dried over MgS04, filtered, and concentrated. The crude product was purified by column chromatography (40 g of silica, 10 to 40%> acetone in hexanes) to afford tert-butyl (5-(((35)-3-(l-propyn-l-yl)-4-(4-(2-(trifluoromethyl)-2-oxiranyl)phenyl)- 1 -piperazinyl)sulfonyl)-2-pyridinyl)carbamate (0.240 g) as an off-white solid.
1H NMR (400MHz, CDC13) δ ppm 8.66 (dd, J= 0.6, 2.3 Hz, 1 H), 8.20 – 8.10 (m, 1 H), 8.04 (dd, J= 2.2, 8.9 Hz, 1 H), 7.63 (s, 1 H), 7.41 (d, J= 8.6 Hz, 2 H), 6.94 (d, J= 8.8 Hz, 2 H), 4.42 (d, J= 2.2 Hz, 1 H), 3.89 – 3.67 (m, 2 H), 3.38 (d, J = 5.3 Hz, 3 H), 2.97 – 2.83 (m, 2 H), 2.80 – 2.60 (m, 1 H), 1.78 (dd, J= 0.8, 2.0 Hz, 3 H), 1.55 (s, 9 H). m/z (ESI, +ve ion) 567.2 (M+H)+.
ALTERNATIVE ROUTE TO 2-(4-BROMOPHENYL)-3,3,3-TRIFLUORO-l,2-PROPANEDIOL (INTERMEDIATE D STEP 2):
F3
![]()
step 1
STEP 1 : 2-(4-BROMOPHENYL)-2-(TRIFLUOROMETHYL)OXIRANE
To a flame-dried, 50-mL, round-bottomed flask was added potassium t-butoxide (0.450 g, 4.01 mmol, Sigma- Aldrich, St. Louis, MO), DMSO (5.0 mL) and trimethylsulfoxonium iodide (1.00 g, 4.54 mmol, Sigma- Aldrich, St. Louis, MO). The resulting mixture was stirred at rt for 40 min. To this reaction mixture was added l-(4-bromophenyl)-2,2,2-trifluoroethanone (1.0 g, 4.0 mmol, Matrix Scientific, Columbia, SC) in DMSO (5.0 mL) dropwise via an addition funnel. The reaction mixture was stirred at rt for 30 min then quenched with water (1 mL) and partitioned between EtOAc (70 mL) and water (30 mL). The organic layer was washed with water (4 x 30 mL), dried over MgS04, filtered, and concentrated. The crude product was purified by column chromatography (40 g of silica, 10 to 20% acetone in hexanes) to afford 2-(4-bromophenyl)-2-(trifluoromethyl)oxirane (0.610 g) as a pale-yellow liquid.
STEP 2: 2-(4-BROMOPHENYL)-3,3,3-TRIFLUORO-l,2-PROPANEDIOL
To a 20-mL vial was added 2-(4-bromophenyl)-2-(trifluoromethyl)oxirane (0.200 g, 0.750 mmol), dioxane (2.0 mL), and water (3.0 mL). The resulting mixture was heated at 85 °C for 24 h. The reaction mixture was cooled to rt and extracted with EtOAc (3 x 50 mL). The combined organic layers were dried over MgS04, filtered and concentrated. The crude product was purified by column chromatography (40 g of silica, 10 to 30% acetone in hexanes) to afford 2-(4-bromophenyl)-3,3,3-trifluoro-l,2-propanediol (2.0 g) as a white solid.
INTERMEDIATE E: BENZYL (3S)-3-(l-PROPYN-l-YL)-l-PIPERAZINECARBOXYLATE
-Cbz ![]()
STEP 1 : 4-BENZYL 1 – TER Γ-BUT YL 2-0X0-1,4-PIPERAZINEDICARBOXYLATE
A 2-L Erlenmeyer flask was charged with 2-piperazinone (36.5 g, 364 mmol, Sigma-Aldrich, St. Louis, MO), sodium carbonate (116 g, 1090 mmol, J. T. Baker, Philipsburg, NJ), 600 mL of dioxane, and 150 mL of water. To this was slowly added benzyl chloroformate (62.1 g, 364 mmol, Sigma-Aldrich, St. Louis, MO) at rt over 20 min. After the addition was complete, the mixture was stirred for 2 h and then diluted with water and extracted with EtOAc (2 L). The combined organic extracts were dried (MgS04), filtered, and concentrated to give a white solid. To this solid was added 500 mL of DCM, triethylamine (128 mL, 911 mmol, Sigma-Aldrich, St. Louis, MO), DMAP (4.45 g, 36.4 mmol, Sigma-Aldrich, St. Louis, MO), and di-tert-butyl dicarbonate (119 g, 546 mmol, Sigma-Aldrich, St. Louis, MO). After stirring at room temperature for 1 h, the mixture was diluted with water and the organics were separated. The organics were dried (MgS04), filtered, and concentrated to give a brown oil. To this oil was added 100 mL of DCM followed by 1 L of hexane. The resulting white solid was collected by filtration to give 4-benzyl 1-tert-butyl 2-oxo-l,4-piperazinedicarboxylate (101 g).
STEP 2: BENZYL (2-((7¾’i?J,-BUTOXYCARBONYL)AMINO)ETHYL)(2-OXO-3 -PENT YN- 1 – YL)C ARB AMATE
A 150-mL round-bottomed flask was charged with 4-benzyl 1-tert-butyl 2-oxo- 1 ,4-piperazinedicarboxylate (1.41 g, 4.22 mmol) and THF (5 mL). 1-Propynylmagnesium bromide (0.5 M in THF, 20.0 mL, 10.0 mmol, Sigma-Aldrich, St. Louis, MO) was added at 0 °C slowly. The mixture was stirred at 0 °C for 2 h. Saturated aqueous NH4C1 (40 mL) was added and the aqueous phase was extracted with EtOAc (200 mL, then 2 x 100 mL). The combined organic phases were dried over sodium sulfate, filtered and concentrated under a vacuum. The crude product was purified by column chromatography (50 g of silica, 0 to 50% EtOAc in hexanes) to afford benzyl (2- tert-butoxycarbonyl)amino)ethyl)(2-oxo-3-pentyn-l-yl)carbamate (1.55 g) as a clear oil.
STEP 3: BENZYL 3-(l-PROPYN-l-YL)-l-PIPERAZINECARBOXYLATE
A 3-L round-bottomed flask was charged with 2-((tert-butoxycarbonyl)amino)ethyl)(2-oxo-3-pentyn-l-yl)carbamate (82.17 g, 219 mmol) and 300 mL of DCM. After cooling to -10 °C, TFA (169 mL, 2200
mmol) was added and the resulting dark solution was stirred at rt for 15 min.
Sodium triacetoxyborohydride (186 g, 878 mmol, Sigma- Aldrich, St. Louis, MO) was then added portion- wise over 10 min. After 2 h, the mixture was
concentrated, diluted with EtOAc (1 L), and neutralized with 5 N NaOH. The layers were separated and the organic extracts were washed with brine, dried (MgS04), filtered and concentrated. The resulting orange oil was purified via column chromatography (750 g of silica gel, 0 to 4.5 % MeOH/DCM) to give benzyl 3 -(l-propyn-l-yl)-l -piperazmecarboxylate (43.67 g) as a brown foam.
STEP 4: 4-BENZYL 1 – TER Γ-BUT YL 2-(l -PROP YN-l-YL)- 1,4-PIPERAZINEDICARBOXYLATE
A 20-mL vial was charged with benzyl 3-(l-propyn-l-yl)-l-piperazinecarboxylate (0.616 g, 2.38 mmol), di-tert-butyl dicarbonate (0.979 g, 4.49 mmol, Sigma-Aldrich, St. Louis, MO), DMAP (0.0287 g, 0.235 mmol, Sigma-Aldrich, St. Louis, MO), TEA (0.90 mL, 6.5 mmol) and DCM (8 mL). The mixture was stirred at rt for 30 min. The reaction mixture was partitioned between water (20 mL) and EtOAc (20 mL). The aqueous phase was extracted with EtOAc (20 mL). The organic phase was washed with saturated aqueous sodium chloride (40 mL), dried over sodium sulfate, filtered, and concentrated under a vacuum. The crude product was purified by column chromatography (25 g of silica, 0 to 50% EtOAc in hexanes) to afford 4-benzyl 1-tert-butyl 2-(l-propyn-l-yl)-l,4-piperazinedicarboxylate (0.488 g) as a colorless oil.
STEP 5: 4-BENZYL 1 – TER Γ-BUT YL (2S)-2-( 1 -PROP YN-l-YL)- 1,4-PIPERAZINEDICARBOXYLATE
The individual enantiomers of 4-benzyl 1-tert-butyl 2-(l-propyn-l-yl)-1 ,4-piperazinedicarboxylate were isolated using chiral SFC. The method used was as follows: Chiralpak® ADH column (Daicel Inc., Fort Lee, NJ) (30 x 250 mm, 5 μιη) using 12% ethanol in supercritical C02 (total flow was 170 mL/min).
This separated the two enantiomers with enantiomeric excesses greater than 98%. The first eluting peak was subsequently identified as 4-benzyl 1-tert-butyl (2S)-2-(l-propyn-l-yl)-l,4-piperazinedicarboxylate and used in the next step.
STEP 6: BENZYL (3S)-3-(l-PROPY -l-YL)-l-PIPERAZINECAPvBOXYLATE
A 100-mL round-bottomed flask was charged with 4-benzyl 1-tert-butyl (25)-2-(l-propyn-l-yl)-l,4-piperazinedicarboxylate (0.145 g, 0.405 mmol), TFA (1.0 mL, 13 mmol) and DCM (2 mL). The mixture was stirred at rt for 40 min. The mixture was concentrated and solid NaHC03 was added followed by saturated aqueous NaHC03. The aqueous phase was extracted with EtOAc (2 x 20 mL). The combined organic phases were washed with IN NaOH (40 mL), saturated aqueous NaHC03 (40 mL), water (40 mL) and saturated aqueous sodium chloride (40 mL). The organic phase was dried over sodium sulfate, filtered, and concentrated under a vacuum to afford benzyl (35)-3-(l-propyn-l-yl)-l-piperazinecarboxylate (0.100 g) as a pale yellow clear oil which solidified upon standing to give a pale yellow solid.
1H NMR (400MHz, MeOD) δ ppm 7.47 – 7.13 (m, 5 H), 5.27 – 5.00 (m, 2 H), 3.88 – 3.58 (m, 3 H), 3.48 – 3.33 (m, 2 H), 3.22 – 3.02 (m, 1 H), 2.89 – 2.63 (m, 1 H), 1.80 (s, 3 H). m/z (ESI, +ve ion) 259.1 (M+H)+.
XAMPLE 23: 5-(((3S)-3-(l-PROPYN-l-YL)-4-(4-(l,2,2,2-TETRAFLUORO-1 -(TRIFLUOROMETHYL)ETHYL)PHENYL)- 1 -PIPERAZINYL)SULFONYL)-2-PYRIDIN AMINE
![]()
STEP 1 : 2-(4-((2S)-4-BENZYL-2-(l-PROPYN-l-YL)-l-PIPERAZINYL)PHENYL)-1 , 1 ,1 ,3,3,3-HEXAFLUORO-2-PROPANOL
A 20-mL vial was charged with (3S)-l-benzyl-3-(l-propyn-l-yl)piperazine (2.143 g, 10 mmol, Intermediate B), 2-(4-bromophenyl)-1,1,1, 3,3, 3-hexafluoropropan-2-ol (3.09 g, 11.5 mmol, Bioorg. Med. Chem. Lett. 2002, 12, 3009), sodium 2-methylpropan-2-olate (1.92 g, 20.0 mmol, Sigma-Aldrich, St. Louis, MO), dioxane (5 mL), RuPhos palladacycle (0.364 g, 0.500 mmol, Strem Chemical Inc., Newburyport, MA), and RuPhos (0.233 g, 0.500 mmol, Strem Chemical Inc., Newburyport, MA). The vial was sealed and heated at 100 °C for 1 h. The mixture was allowed to cool to rt, and diluted with water and extracted with EtOAc. The combined organic phases were dried over sodium sulfate, filtered and concentrated under a vacuum to give a solid that was purified by silica gel column chromatography (0 to 40% EtOAc in hexanes) to afford 2-(4-((2S)-4-benzyl-2-( 1 -propyn- 1 -yl)- 1 -piperazinyl)phenyl)- 1,1,1,3,3,3-hexafluoro-2-propanol (1.75 g) as a slightly yellow oil.
STEP 2: l,l,l,3,3,3-HEXAFLUORO-2-(4-((2S)-2-(l-PROPYN-l-YL)-l-PIPERAZINYL)PHENYL)-2-PROPANOL
A 250 mL round-bottomed flask was charged with 2-(4-((2S)-4-benzyl-2-( 1 -propyn- 1 -yl)- 1 -piperazinyl)phenyl)- 1,1,1 ,3 ,3 ,3-hexafluoro-2-propanol (1.75 g, 4.35 mmol), potassium carbonate (2.40 g, 17.4 mmol, Sigma-Aldrich, St. Louis, MO), CH2CI2 (25 mL), and 1-chloroethyl chlorocarbonate (1.88 mL, 17.4 mmol, Sigma-Aldrich, St. Louis, MO). After 30 min at rt, the reaction was filtered and the filtrate was concentrated. To the resulting oil was added MeOH (25 mL). This mixture was heated at 75 °C for 1.5 h then concentrated. The residue was triturated with diethyl ether to give l,l,l,3,3,3-hexafluoro-2-(4-((2S)-2-(l-propyn-l-yl)-l-piperazinyl)phenyl)-2-propanol (1.44 g) as a white solid.
STEP 3: TERT-BUTYL (5-(((3S)-3-(l-PROPYN-l-YL)-4-(4-(2,2,2-TRIFLUORO- 1 -HYDROXY- 1 -(TRIFLUOROMETHYL)ETHYL)PHENYL)- 1 -PIPERAZINYL)SULFONYL)-2-PYRIDINYL)CARBAMATE
A 250-mL round-bottomed flask was charged with 1,1,1,3,3,3-hexaf uoro-2-(4-((2S)-2-( 1 -propyn- 1 -yl)- 1 -piperazinyl)phenyl)-2-propanol (18.9 g, 51.6 mmol) and DCM (150 mL) and cooled to 0 °C. TEA was added (14.4 mL, 103 mmol, Sigma-Aldrich, St. Louis, MO) followed by tert-butyl (5- (chlorosulfonyl)pyridin-2-yl)carbamate (15.9 g, 54.2 mmol, Intermediate A) portionwise. After 10 min, the reaction mixture was diluted with water (100 mL) and the organic layer was separated, dried over Na2S04, filtered and concentrated under a vacuum to give a solid that was purified by silica gel column
chromatography (0 to 50% EtO Ac in hexanes) to afford tert-butyl (5 -(((3 S)-3 -( 1 -propyn- 1 -yl)-4-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 -(trifluoromethyl)ethyl)phenyl)- 1 -piperazinyl)sulfonyl)-2-pyridinyl)carbamate (19.9 g) as a tan foam.
STEP 4: 5-(((3S)-3-(l-PROPYN-l-YL)-4-(4-(l,2,2,2-TETRAFLUORO-l- (TRIFLUOROMETHYL)ETHYL)PHENYL)- 1 -PIPERAZINYL)SULFONYL)-2-PYRIDIN AMINE
A 500-mL round-bottomed flask was charged with tert-butyl (5-(((3S)-3-(1 -propyn- 1 -yl)-4-(4-(2,2,2-trifluoro- 1 -hydroxy- 1 – (trifluoromethyl)ethyl)phenyl)-l-piperazinyl)sulfonyl)-2-pyridinyl)carbamate (19.7 g, 31.6 mmol) and DCM (300 mL) and cooled to 0 °C.
(Diethylamino)sulfur trifluoride (4.18 mL, 31.6 mmol, Matrix Scientific, Columbia, SC) was added, and after 10 min, the reaction was diluted with water (250 mL) and DCM (200 mL). The organic layer was separated, dried over
Na2S04, filtered and concentrated under a vacuum. The resultant foam was taken up in DCM (200 mL) and cooled to 0 °C. TFA (100 mL, 1298 mmol) was added and the reaction mixture was warmed to rt for 1.5 h. The reaction was then re-cooled to 0 °C and solid sodium bicarbonate was added slowly until gas evolution ceased. The mixture was diluted with water (250 mL) and DCM (300 mL) and the organic layer was separated, dried over Na2S04, filtered and concentrated under a vacuum to give a solid that was purified by silica gel column chromatography (0 to 100% EtOAc in hexanes) to afford 5-(((3S)-3-(l-propyn- 1 -yl)-4-(4-( 1 ,2,2,2-tetrafluoro- 1 -(trifluoromethyl)ethyl)phenyl)- 1 -piperazinyl)sulfonyl)-2-pyridinamine (11.05 g) as a single enantiomer.
1H NMR (400MHz, CD3OD) δ ppm 8.31 (d, J= 2.2 Hz, 1 H), 7.74 (dd, J= 2.4, 8.9 Hz, 1 H), 7.47 (d, J = 8.8 Hz, 2 H), 7.12 (d, J = 9.0 Hz, 2 H), 6.63 (d, J= 8.8 Hz, 1 H), 4.76-4.70 (m, 1 H), 3.76 (dd, J= 1.9, 11.2 Hz, 2 H), 3.66 – 3.52 (m, 1 H), 3.29 – 3.20 (m, 1 H), 2.79 – 2.72 (m, 1 H), 2.66 – 2.53 (m, 1 H), 1.76 (d, J = 2.2 Hz, 3 H). m/z (ESI, +ve ion) 525.2 (M+H)+. GK-GKRP IC50 (Binding) = 0.187 μΜ.
PAPER
Small Molecule Disruptors of the Glucokinase–Glucokinase Regulatory Protein Interaction: 2. Leveraging Structure-Based Drug Design to Identify Analogues with Improved Pharmacokinetic Profiles
David J. St. JeanJr.*†, Kate S. Ashton†, Michael D. Bartberger‡, Jie Chen∥, Samer Chmait‡, Rod Cupples§, Elizabeth Galbreath⊥, Joan Helmering§, Fang-Tsao Hong†, Steven R. Jordan‡, Longbin Liu†, Roxanne K. Kunz†, Klaus Michelsen‡, Nobuko Nishimura†, Lewis D. Pennington†, Steve F. Poon†, Darren Reid#, Glenn Sivits§, Markian M. Stec†, Seifu Tadesse†, Nuria Tamayo†, Gwyneth Van⊥, Kevin C. Yang†, Jiandong Zhang‡, Mark H. Norman†, Christopher Fotsch†, David J. Lloyd§, and Clarence Hale§
†Department of Therapeutic Discovery—Medicinal Chemistry, ‡Department of Therapeutic Discovery—Molecular Structure and Characterization, §Department of Metabolic Disorders, ∥Department of Pharmacokinetics and Drug Metabolism, ⊥Department of Pathology, #Department of Pharmaceutics Amgen, Inc., One Amgen Center Drive, Thousand Oaks, California, 91320 and 360 Binney Street, Cambridge, Massachusetts, 02142, United States
J. Med. Chem., 2014, 57 (2), pp 325–338
DOI: 10.1021/jm4016747
In the previous report, we described the discovery and optimization of novel small molecule disruptors of the GK-GKRP interaction culminating in the identification of 1 (AMG-1694). Although this analogue possessed excellent in vitro potency and was a useful tool compound in initial proof-of-concept experiments, high metabolic turnover limited its advancement. Guided by a combination of metabolite identification and structure-based design, we have successfully discovered a potent and metabolically stable GK-GKRP disruptor (27, AMG-3969). When administered to db/db mice, this compound demonstrated a robust pharmacodynamic response (GK translocation) as well as statistically significant dose-dependent reductions in fed blood glucose levels.
2-(4-((2S)-4-((6-Amino-3-pyridinyl)sulfonyl)-2-(1-propyn-1-yl)-1-piperazinyl)phenyl)-1,1,1,3,3,3-hexafluoro-2-propanol (27)
1H NMR (400 MHz, CDCl3) δ 8.48 (d, J = 2.3 Hz, 1 H), 7.77 (dd, J = 2.5, 8.8 Hz, 1 H), 7.57 (d, J = 8.8 Hz, 2 H), 6.95 (d, J = 9.2 Hz, 2 H), 6.52 (d, J = 8.8 Hz, 1 H), 4.94 (s, 2 H), 4.44 (br s, 1 H), 3.82–3.71 (m, 2 H), 3.58–3.33 (m, 3 H), 2.81 (dd, J = 3.2, 11.1 Hz, 1 H), 2.67 (dt, J = 3.9, 11.0 Hz, 1 H), 1.78 (d, J = 2.2 Hz, 3 H).
m/z (ESI, +ve ion) 523.2 (M + H)+.
REFERENCES
St Jean, D.J. Jr.; Ashton, K.; Andrews, K.; et al.
Small molecule disruptors of the glucokinase-glucokinase regulatory protein (GK-GKRP) interaction
34th Natl Med Chem Symp (May 18-21, Charleston) 2014, Abst 4
Small molecule disruptors of the GK-GKRP interaction as potential antidiabetics
247th Am Chem Soc (ACS) Natl Meet (March 16-20, Dallas) 2014, Abst MEDI 214
Use of non-traditional conformational restriction in the design of a novel, potent, and metabolically stable series of GK-GKRP inhibitors
248th Am Chem Soc (ACS) Natl Meet (August 10-14, San Francisco) 2014, Abst MEDI 267
Small molecule inhibitors for glucokinase-glucokinase regulatory protein (GK-GKRP) binding: Optimization for in vivo target assessment of type II diabetes
248th Am Chem Soc (ACS) Natl Meet (August 10-14, San Francisco) 2014, Abst MEDI 268
![]()
MAKING CONNECTIONS Aleksandra Baranczak (right), a fourth-year grad student in Gary A. Sulikowski’s lab at Vanderbilt University, discusses her efforts to synthesize the core of the diazo-containing natural product lomaiviticin A with Kate Ashton, a medicinal chemist at Amgen
Dr. Kate Ashton
![Mark Norman]()
Mark Norman
![Michael Bartberger]()
Michael Bartberger
![Chris Fotsch]()
Chris Fotsch
![David St. Jean]()
David St. Jean
![Klaus Michelsen]()
Klaus Michelsen
///////////1361224-53-4, AMGEN, AMG 3969, Type 2 Diabetes, PRECLINICAL
O=S(=O)(c1ccc(N)nc1)N2C[C@H](C#CC)N(CC2)c3ccc(cc3)C(O)(C(F)(F)F)C(F)(F)F
Filed under:
Preclinical drugs,
Uncategorized Tagged:
1361224-53-4,
amg 3969,
amgen,
preclinical,
TYPE 2 DIABETES ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

























































 was synthesized as outline in Scheme I.
was synthesized as outline in Scheme I.



 Center for Molecular Discovery (CMD) Director John Porco and members of the CMD lab team.
Center for Molecular Discovery (CMD) Director John Porco and members of the CMD lab team.






 Randolph A. Escobar
Randolph A. Escobar











![1,2-bis[4-(4-nitrophenyl)piperazin-1-yl]ethanone.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=2867018&t=l "A structure of 1,2-bis[4-(4-nitrophenyl)piperazin-1-yl]ethanone")
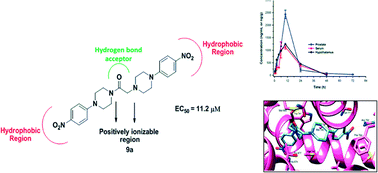






















 DDD498
DDD498











































































































































 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..



 amcrasto@gmail.com
amcrasto@gmail.com